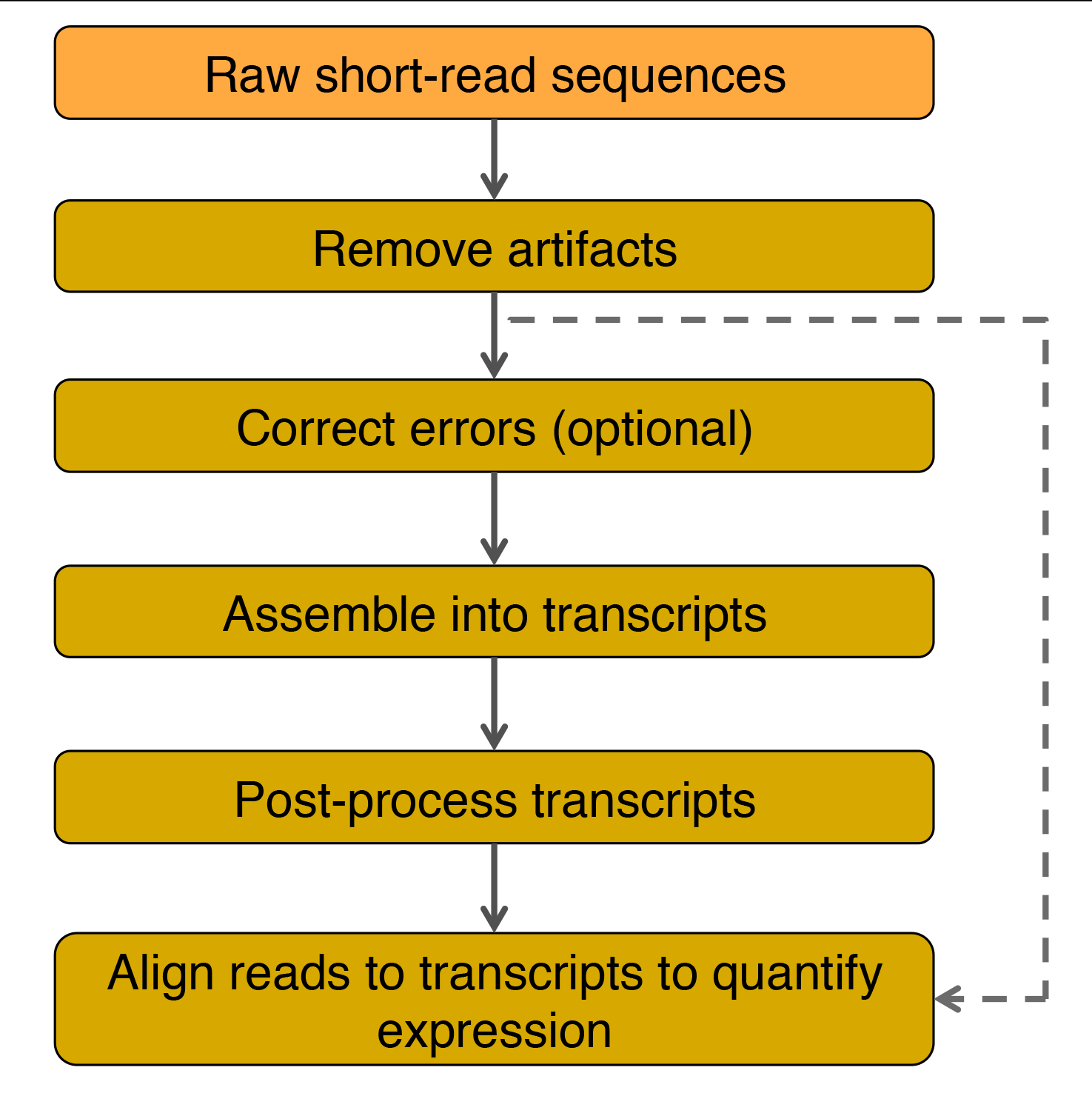
Flow chart for transcriptome assembly and quantification of gene expression. Adapted from Martin and Wang (2011).
Data Analysis
Analysis of raw short reads can be separated into two main steps: pre-processing the raw data and transcriptome assembly (see Martin and Wang, 2011 for details).
Pre-proceesing raw short-read sequences
- Removal of the following artifacts will improve the sequence read quality.
- Sequencing adapters from failed or short DNA insertions during library construction.
- Low-complexity reads and near-identical reads arise from PCR amplification.
- If identities are known, rRNA and other RNA contamimants should be removed to improve assembly speed.
- Sequencing errors can be removed or corrected by analyzing the quality score and/or the k-mer frequency (the number of times that each k-length oligonucleotide appears in a sequence).
- Low quality scores indicate possible sequencing errors.
- Low frequency for a k-mer indicates a possible sequencing error or a low abundance transcript.